Statistical Methods Development for 3D Genomics Data Analysis
Overview: Our project focuses on developing statistical methods and computational tools for analyzing three-dimensional genome structure data. With the emergence of Hi-C and other 3C-derived technologies, we can now investigate chromatin interactions at unprecedented resolution, from megabase to kilobase scales. Our research addresses key challenges in processing and analyzing these complex datasets through innovative statistical approaches and efficient computational implementations.
Motivation: The three-dimensional organization of chromatin plays a crucial role in gene regulation and disease development. Our research addresses several fundamental challenges in 3D genomics through multiple innovative approaches:

mHi-C: A probabilistic framework for utilizing multi-mapping reads in Hi-C data analysis
- Recovers ~20% additional sequencing depth typically discarded
- Refines chromatin domain boundaries
- Identifies novel promoter-enhancer interactions
FreeHi-C & FreeHi-C SpikeIn: Empirical simulation tools for Hi-C data
- Generates realistic Hi-C contact matrices
- Enables power analysis and method development
- Provides user-controlled noise levels
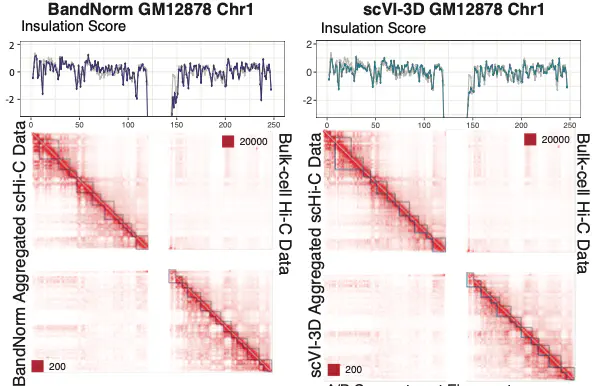
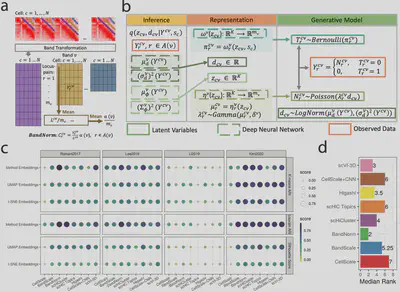
BandNorm & scVI-3D: Advanced tools for single-cell Hi-C analysis
- Addresses technical biases in scHi-C data
- Enables robust cell-type identification
- Facilitates rare cell type analysis
The project’s success has led to improved understanding of:
- Long-range gene regulation mechanisms
- Disease-associated genetic variants
- Chromatin domain structure in cancer
- Cell-type specific genome organization
Our long-term goal is to continue developing robust computational methods that advance our understanding of 3D genome organization and its role in human disease.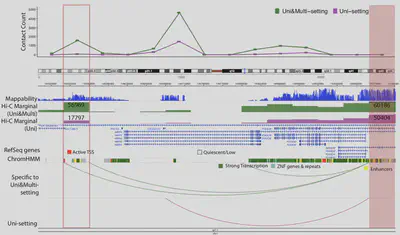
Publications
+: co-corresponding author *: co-first author
- Zheng Y+, Shen S+, Keleş S. Normalization and De-noising of Single-cell Hi-C Data with BandNorm and scVI-3D. Accepted by Genome Biology. 2022.
- Cheng J, Clayton J, Acemel R, Zheng Y, Taylor R, Keleş S, Harley J, Quail E, Gómez-Skarmeta J and Ulgiati D. Regulatory architecture of the RCA gene cluster captures an intragenic TAD boundary and enhancer elements in B cells. Frontiers in Immunology, section B Cell Biology. 2022.
- Zheng Y, Zhou P, Keleş S. FreeHi-C Spike-in Simulations for Benchmarking Differential Chromatin Interaction Detection. Methods. 2021.
- Huang K, Wu Y, Shin J, Zheng Y, Siahpirani A, Lin Y, Ni Z, Chen J, You J, Keleş S, Wang D, Roy S, Lu Q. Transcriptome-wide transmission disequilibrium analysis identifies novel risk genes for autism spectrum disorder. PLOS Genetics. 2021.
- Zheng Y, Keleş S. FreeHi-C simulates high-fidelity Hi-C data for benchmarking and data augmentation. Nature Methods. 2020.
- The ENCODE Project Consortium, et al. Expanded Encyclopedias of DNA Elements in the Human and Mouse Genomes. Nature. 2020 .
- The ENCODE Project Consortium, Snyder, M.P., Gingeras, T.R., Moore, J.E., Weng, Z., Gerstein, M.B., Ren, B., Hardison, R.C., Stamatoyannopoulos, J.A., Graveley, B.R., Feingold, E.A. and Pazin, M.J. Perspectives on ENCODE. Nature. 2020.
- Zheng Y, Ay F, Keleş S. Generative modeling of multi-mapping reads with mHi-C advances analysis of Hi-C studies. eLife. 2019.
Ye Zheng
Assistant Professor, PI
Research interests include Multi-omics, Statistical Modeling, Computational Biology, Cancer Research