mHiC
 mHiC workflow
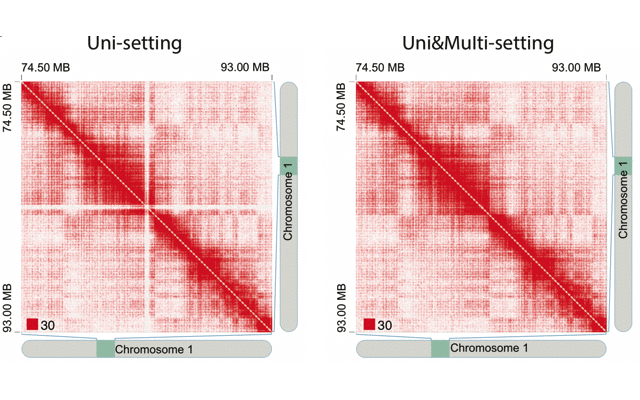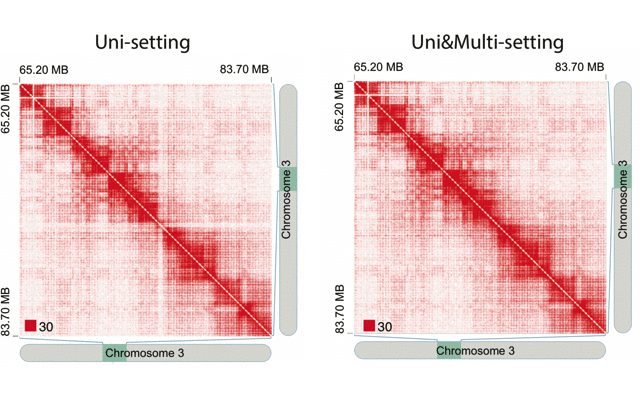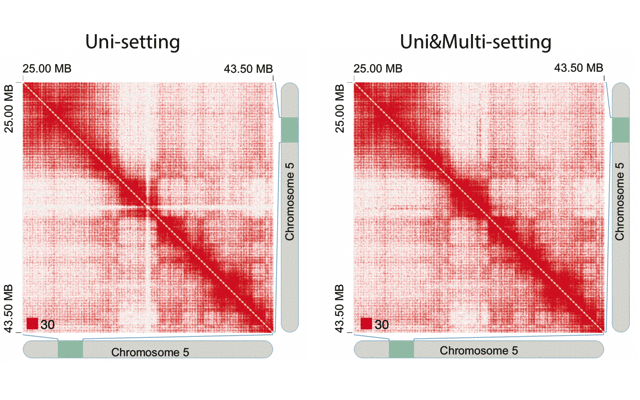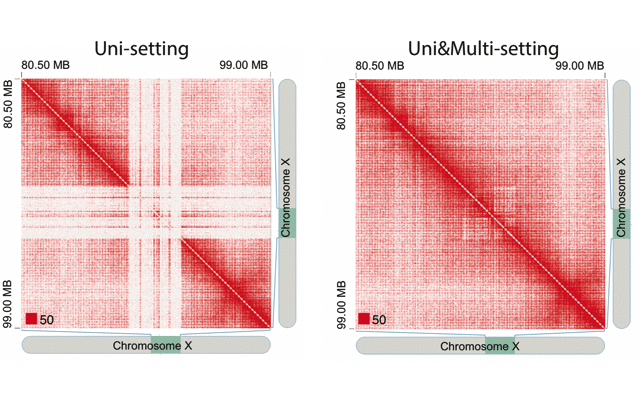
mHiC workflowOverview: mHi-C (multi-mapping strategy for Hi-C data) is an innovative pipeline designed to leverage reads that align to multiple genomic positions in Hi-C analysis. By implementing a probabilistic framework for assigning multi-mapping reads, mHi-C significantly improves the utilization of sequencing data and enhances the detection of chromatin interactions.
Key Features:
Multi-mapping Read Processing:
- Probabilistic assignment of ambiguous reads
- Statistical modeling of mapping locations
- Comprehensive read classification
- Improved data utilization
Flexible Analysis Pipeline:
- Modular step-by-step workflow
- Independent script organization
- User-defined parameter settings
- Customizable implementation options
Performance Optimization:
- C/C++ accelerated computing
- Parallel processing support
- High-performance computing integration
- Memory-efficient implementation
Quality Control and Validation:
- Statistical significance assessment
- Resolution-specific analysis
- Comprehensive quality metrics
- Robust validation methods
Applications: mHiC has been successfully applied to:
- Analyze repetitive genomic regions
- Improve chromatin interaction detection
- Enhance Hi-C data utilization
- Generate high-resolution contact maps
- Study complex genomic architectures
The package is implemented in Python with C/C++ acceleration for computationally intensive components, providing comprehensive documentation and tutorials to support researchers in the fields of genomics and chromatin biology.
Ye Zheng
Assistant Professor, PI
Research interests include Multi-omics, Statistical Modeling, Computational Biology, Cancer Research